
FDA Issues Guidance on Applying Human Factors and Usability Engineering to Medical Devices
FDA has issued guidance on applying human factors and usability engineering to medical devices, focusing on user interface design, use-related risk management, critical tasks and human factors validation testing.

FDA Announces Opening of FY2027 MDUFA Small Business Determination Window
FDA opens the FY2027 MDUFA Small Business Request window on August 1, 2026. Learn eligibility rules, user fee discounts, and submission steps.
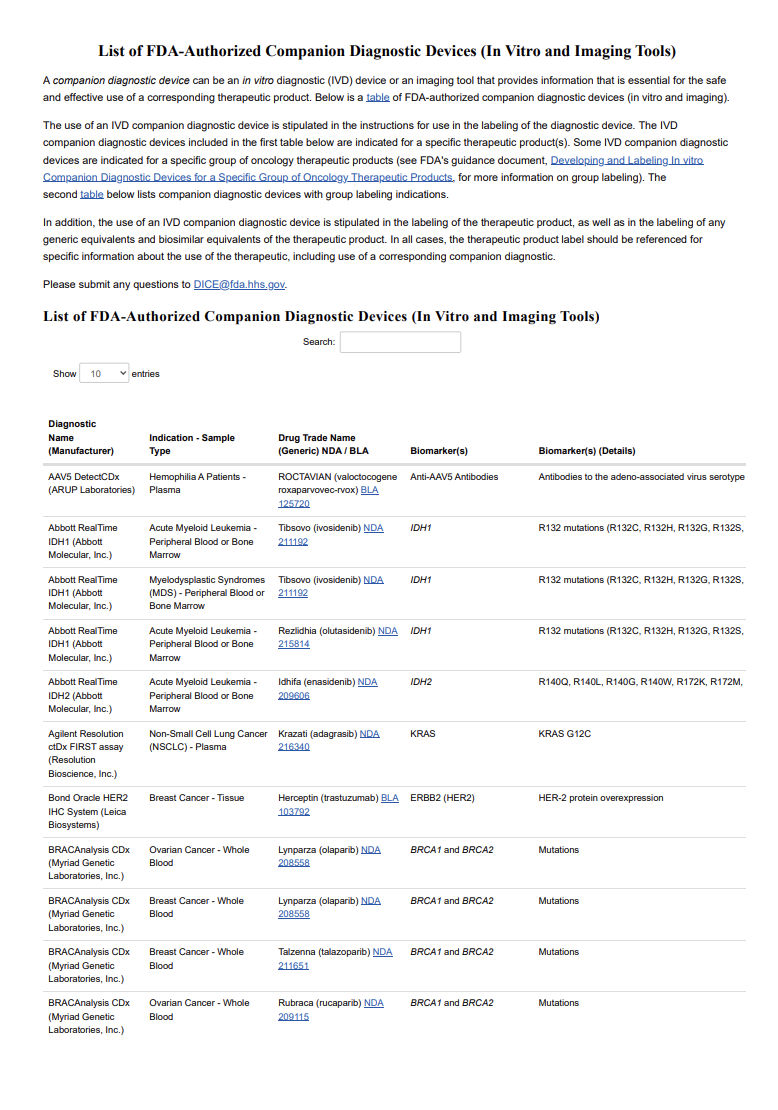
FDA Updates Authorized Companion Diagnostic Devices List to 226 Entries
FDA updates its list of authorized companion diagnostic devices to 226 entries, covering NGS assays, liquid biopsies, group labeling, and key biomarkers.

2026 MDSAP Forum Outcome Statement Highlights Global Expansion and EU Alignment
The MDSAP Regulatory Authority Council releases the 2026 Forum Outcome Statement, detailing 2025 performance, MDO pilot results, and EU MDR alignment.

FDA Issues Draft Guidance on Biocompatibility Testing Under the ASCA Program
FDA has issued draft guidance on biocompatibility testing of medical devices under the ASCA Program, covering recognized standards, laboratory accreditation, test methods and premarket submission content.

FDA CBER Updates SOPP on MDUFA User Fee Payments and Billing Activities
FDA CBER has updated SOPP 8704 on MDUFA user fee payments and billing activities, including electronic payments, payment verification, user fee holds, refunds and establishment registration fees.

FDA Updates Guidance on Submitting Next-Generation Sequencing Data to the Division of Antivirals
FDA has updated its technical specifications guidance on submitting next-generation sequencing protocols, data and analyses to the Division of Antivirals.

FDA Announces Upcoming eMDR System Enhancements
The FDA has announced upcoming eMDR system enhancements, including AEMS consolidation, country code requirements and IMDRF adverse event code updates.

FDA Classifies Endoscopic Traction Devices as Class II Medical Devices
FDA classifies the Endoscopic Traction Device as a Class II medical device with Special Controls, setting performance, safety, and labeling requirements applicable to medical device and IVD manufacturers.

FDA Classifies Ingestible Gastrointestinal Blood Detection Capsule as a Class II Medical Device
The FDA has classified the ingestible gastrointestinal blood detection capsule as a Class II medical device, establishing special controls to support the safety and effectiveness of this innovative diagnostic technology.

FDA Classifies Radiological Machine Learning-Based Quantitative Imaging Software with Predetermined Change Control Plans as Class II Devices
The FDA has classified radiological machine learning-based quantitative imaging software with predetermined change control plans as Class II medical devices, establishing special controls to support the safe development and regulatory oversight of AI-enabled imaging software.

FDA Publishes Draft Guidance on Forms FDA 3542a and FDA 3542 for Orange Book Patent Submissions
FDA has published draft guidance on Forms FDA 3542a and FDA 3542, clarifying requirements for Orange Book patent listings, electronic patent submissions, and the management of patent information throughout the medicinal product lifecycle.

FDA Publishes Draft Guidance on Essential Drug Delivery Outputs for Drug Delivery Devices
FDA has published draft guidance on Essential Drug Delivery Outputs (EDDOs) for drug delivery devices and combination products. Learn how the recommendations may impact design controls, verification, validation, and change management activities.

FDA Updates Guidance on Manufacturer Communications with Payors: What Medical Device Companies Need to Know
The FDA has updated its draft guidance on medical device manufacturer communications with payors, health economic information, and investigational products.

FDA Updates eSTAR Program: Human Factors Content Added and Expanded PreSTAR Functionality
The FDA has released eSTAR Version 7.0, integrating new Human Factors content and expanding PreSTAR capabilities. Learn how these changes may affect medical device manufacturers preparing FDA submissions.

FDA Establishes Class II Classification for TENS Devices Intended to Reduce Fibromyalgia Symptoms
FDA has established a new Class II classification with special controls for transcutaneous electrical nerve stimulators (TENS) intended to treat fibromyalgia symptoms, creating a new regulatory pathway for manufacturers.

FDA Reports 1,284 Devices Granted Breakthrough Device Designation as of March 2026
The FDA has reported 1,284 Breakthrough Device designations and 198 market-authorised Breakthrough Devices as of March 2026. Learn what this means for medical device manufacturers and U.S. market access strategies.

FDA Adds Five Additional Unclassified Medical Devices to 510(k) Enforcement Discretion Policy
FDA has updated its guidance on unclassified medical devices, adding five additional device types to its 510(k) enforcement discretion policy. Learn what this means for manufacturers and ongoing FDA compliance obligations.

FDA Adopts ICH M11 CeSHarP Guidance: What Sponsors Need to Know About the Future of Clinical Trial Protocols
The FDA has adopted ICH M11 CeSHarP, introducing a harmonized framework for clinical trial protocols. Discover the impact on sponsors and global studies.

FDA Finalizes Risk-Based Guidance on Human Factors Information in Medical Device Submissions
The FDA has published its final guidance on Human Factors information in medical device submissions, introducing a new risk-based framework and three Human Factors Submission Categories. Learn what manufacturers need to know.