
MHRA Updates Guidance on Submitting Clinical Investigation Proposals for Assessment
MHRA has updated its guidance on submitting clinical investigation proposals, covering GB and Northern Ireland requirements, IRAS submission, supporting documentation, fees, device evidence, software, biological safety and sterilisation.

FDA Issues Updated Guidance on 510(k) Submissions for Hemodialysis Blood Tubing Sets
FDA has issued updated guidance on 510(k) submissions for hemodialysis blood tubing sets, covering device description, performance testing, biocompatibility, sterility, shelf life, packaging and labeling.

IMDRF Publishes Final Document on Predetermined Change Control Plans for Medical Device Software
IMDRF has published final principles on Predetermined Change Control Plans for medical device software, covering PCCP elements, risk-based change control, impact assessment, benefits and challenges.
URL slug sugerido:

Team-NB Calls for Greater Transparency and EUDAMED Access for Notified Bodies
Team-NB has published a position paper calling for broader notified body access to EUDAMED and related regulatory systems to support risk-based surveillance under the MDR and IVDR.

Anvisa Launches New Call for Regulatory Evaluation of Innovative Medical Devices
Anvisa has launched Call Notice 5/2026 to select up to 10 innovative medical device projects for regulatory evaluation and continuous dialogue in Brazil.

FDA Issues Guidance on Applying Human Factors and Usability Engineering to Medical Devices
FDA has issued guidance on applying human factors and usability engineering to medical devices, focusing on user interface design, use-related risk management, critical tasks and human factors validation testing.

Swissmedic Updates Guidance on Export Certificates for Medical Devices
Swissmedic has updated its guidance on export certificates for medical devices, including eligibility, ordering requirements, product lists, CE conformity documentation, swissdamed registration and fees.

TGA Updates Guidance on Selection Criteria for Medical Device Application Audits
The TGA has updated its guidance on how medical device applications are selected for audit before inclusion in the ARTG, including mandatory audits, non-mandatory audit criteria, regulatory reforms, UDI and post-market signals.

MDSAP Updates Audit Approach Document with Expanded Process-Based Guidance
The updated MDSAP Audit Approach document, MDSAP AU P0002.011, provides detailed instructions for conducting process-based MDSAP audits and integrates the former Audit Model and Process Companion documents.

NIST Publishes Generative AI Profile for the AI Risk Management Framework
NIST AI 600-1 provides a Generative AI Profile for the AI Risk Management Framework, identifying key GAI risks and suggested actions to govern, map, measure and manage them.

Team-NB Calls for Broader EUDAMED Transparency to Support Risk-Adaptive Surveillance
Team-NB has highlighted the need for broader notified body access to EUDAMED vigilance, clinical investigation and performance study data to support proportionate risk-adaptive surveillance under the MDR and IVDR.

Team-NB Highlights Risk-Adaptive Surveillance under the MDR and IVDR
Team-NB has presented a proposal for risk-adaptive notified body surveillance under the MDR and IVDR, aiming to reduce unnecessary burden while maintaining patient safety and effective oversight.

ISO Publishes ISO 14155:2026 on Good Clinical Practice for Medical Device Clinical Investigations
ISO 14155:2026 specifies good clinical practice for the design, conduct, recording and reporting of clinical investigations of medical devices involving human subjects.

MHRA Issues 5th Edition Safety Guidelines for Magnetic Resonance Imaging Equipment in Clinical Use
MHRA releases the 5th edition of its Safety Guidelines for Clinical MRI Equipment, updating governance, ASTM F2503-26 standards, RF burns, and implant rules.
ICH Publishes Updated Support Package for ICH M8 eCTD v4.0
ICH M8 Working Group updates eCTD v4.0 support materials, including the Implementation Guide, Controlled Vocabularies, and Q&A documentation.

FDA Announces Opening of FY2027 MDUFA Small Business Determination Window
FDA opens the FY2027 MDUFA Small Business Request window on August 1, 2026. Learn eligibility rules, user fee discounts, and submission steps.
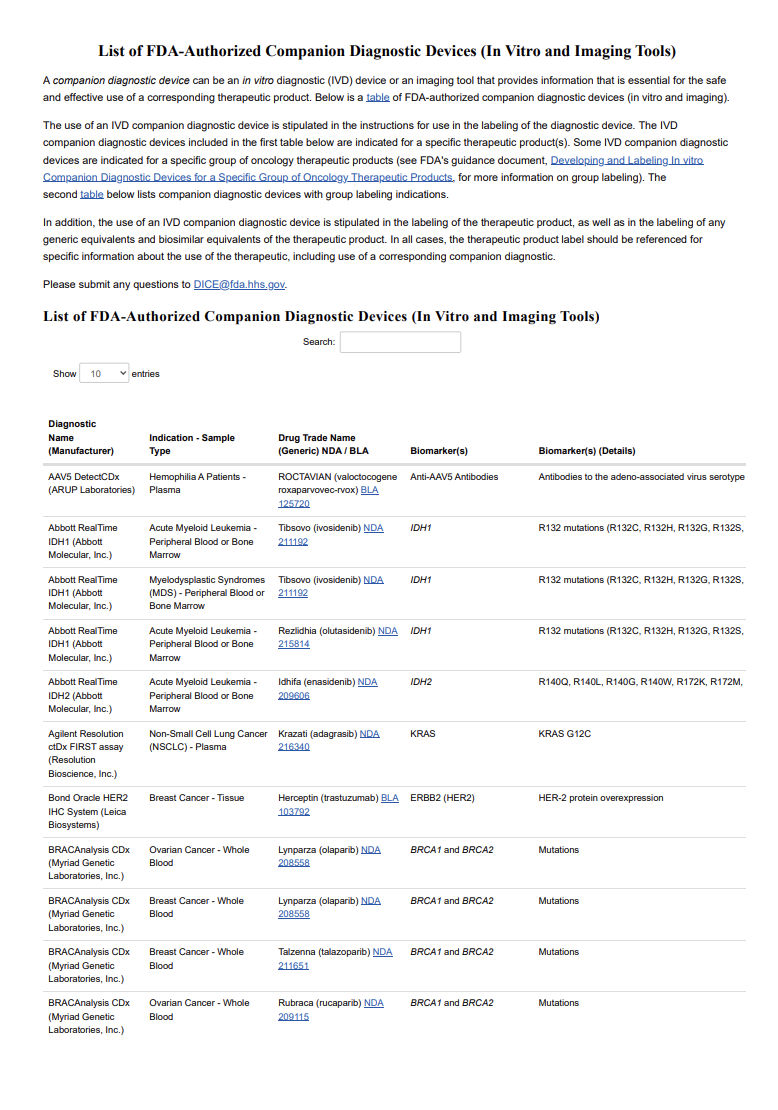
FDA Updates Authorized Companion Diagnostic Devices List to 226 Entries
FDA updates its list of authorized companion diagnostic devices to 226 entries, covering NGS assays, liquid biopsies, group labeling, and key biomarkers.

MHRA Releases AI Airlock Sandbox Phase 2 Programme Report
MHRA releases its AI Airlock Sandbox Phase 2 report, detailing regulatory insights on AI validation, PCCPs, generative AI, and real-world PMS.

2026 MDSAP Forum Outcome Statement Highlights Global Expansion and EU Alignment
The MDSAP Regulatory Authority Council releases the 2026 Forum Outcome Statement, detailing 2025 performance, MDO pilot results, and EU MDR alignment.

FDA Issues Guidance on Cancer Clinical Trial Eligibility: Laboratory Values
FDA issues guidance on cancer clinical trial eligibility, recommending evidence-based laboratory criteria to reduce unnecessary exclusions in oncology studies.