
TGA Updates Guidance on Medical Device Application Audit Selection Criteria
The TGA has updated its guidance on how medical device and IVD applications are selected for audit before inclusion in the ARTG.

TGA Introduces Australia's UDI System with Phased Implementation Through 2029
The TGA has introduced Australia's Unique Device Identification (UDI) system with a phased implementation through 2029. Learn about the implementation timeline, AusUDID requirements, and what medical device and IVD manufacturers need to prepare.

Swissmedic publishes updated swissdamed Playground User Guide
Swissmedic has published Version 3.0 of the swissdamed User Guide – Playground, providing updated instructions for testing actor registration and UDI management before using the production environment.
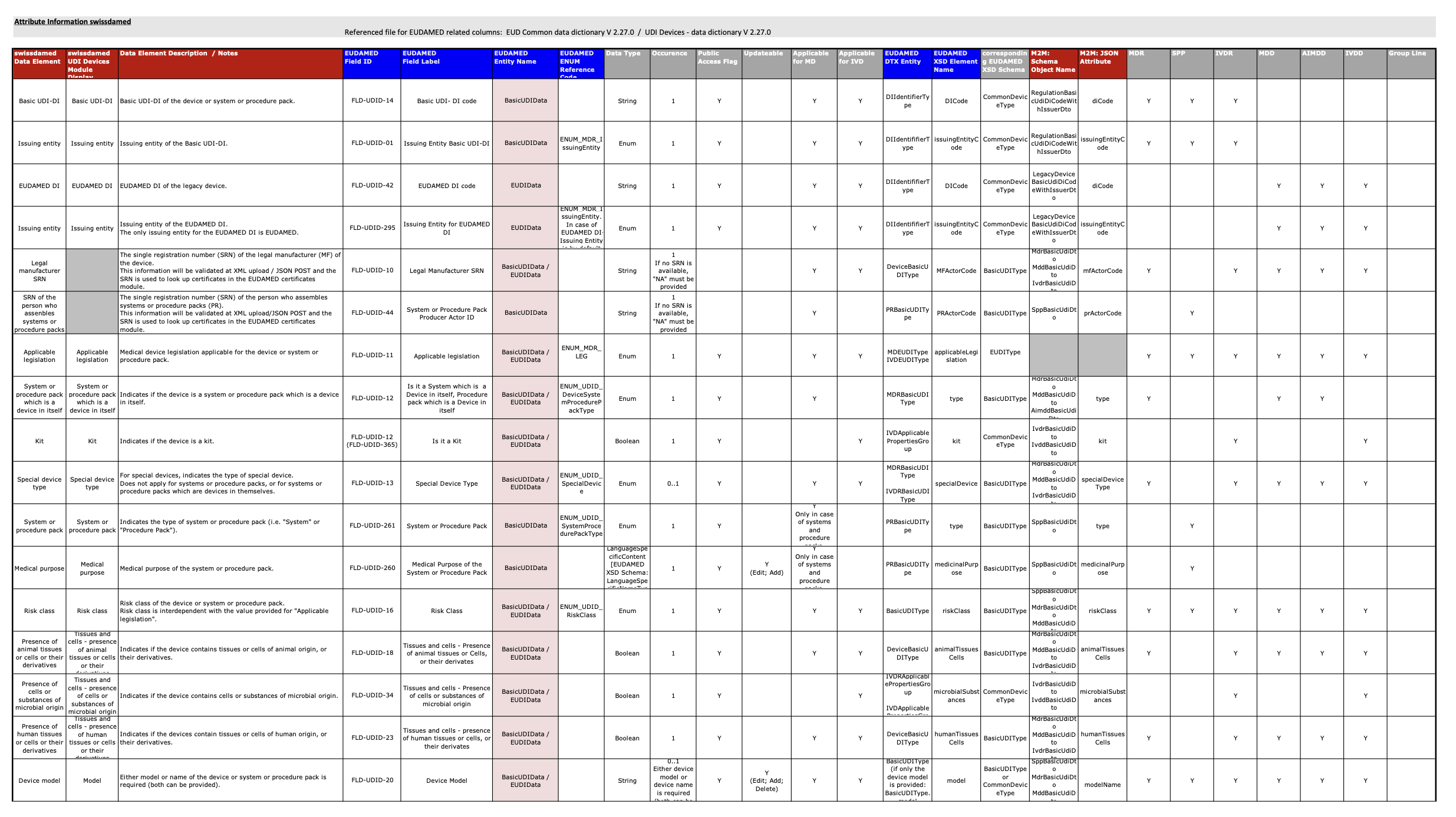
Swissmedic Publishes Version 2.0 of the swissdamed UDI Data Dictionary
Swissmedic has published Version 2.0 of the swissdamed UDI Data Dictionary, introducing new data elements and clarifying requirements for the registration and management of UDI information for medical devices in Switzerland.

Swissmedic Publishes User Guide for the swissdamed UDI Devices Module
Swissmedic has published Version 3.0 of the swissdamed User Guide – UDI Devices Module, introducing new functionalities for device registration and management, including Machine-to-Machine (M2M) processes, manual UDI-DI management and Master UDI-DI requirements.

Swissmedic Publishes swissdamed Business Rules Version 4.0
Swissmedic has published swissdamed Business Rules Version 4.0, introducing new Master UDI-DI requirements, UDI registration rules, and data management obligations for medical device manufacturers in Switzerland.

Australia Updates Guidance on UDI Compliance Timeframes for Medical Devices
The TGA has updated its guidance on UDI compliance timeframes for medical devices in Australia, including clarifications on legacy devices, EU MDR transitions, consignment stock, and Surgical Loan Kits.

TGA Introduces Streamlined UDI Consent-to-Supply Pathway for Non-Compliant Medical Devices
Australia’s TGA has released new guidance on streamlined UDI Consent-to-Supply applications for medical devices that do not meet UDI-related Essential Principles. Learn how the changes affect manufacturers, ARTG entries, timelines, fees, and compliance strategies.

European Commission Publishes Updated EUDAMED UDI Devices User Guide
The European Commission released version 2.27.0 of the EUDAMED UDI Devices User Guide, clarifying UDI registration, EMDN management, packaging levels, and lifecycle updates under MDR and IVDR.
Swissmedic Updates swissdamed Actors User Guide: What Manufacturers and Authorised Representatives Should Know
Swissmedic has updated the swissdamed Actors User Guide (v2.0), introducing AGOV login migration updates, mandate transfer functionality, and clarified actor management requirements for medical device manufacturers and authorised representatives in Switzerland.

Swissmedic Issues Minor Update to Guidance on User Incident Reporting
Swissmedic updates guidance on medical device incident reporting. Key timelines, obligations, and implications for manufacturers explained.

European Commission publishes EUDAMED Release Notes v2.25.2
The European Commission releases EUDAMED v2.25.2 with new validation rules, certificate controls, and API access. Key implications for medical device manufacturers.

MDCG 2025-8 Rev.1: Guidance on Master UDI-DI for Spectacle Devices
MDCG 2025-8 Rev.1 provides guidance on Master UDI-DI implementation for spectacle frames, lenses, and reading spectacles under EU MDR. Learn key requirements, timelines, and assignment rules.
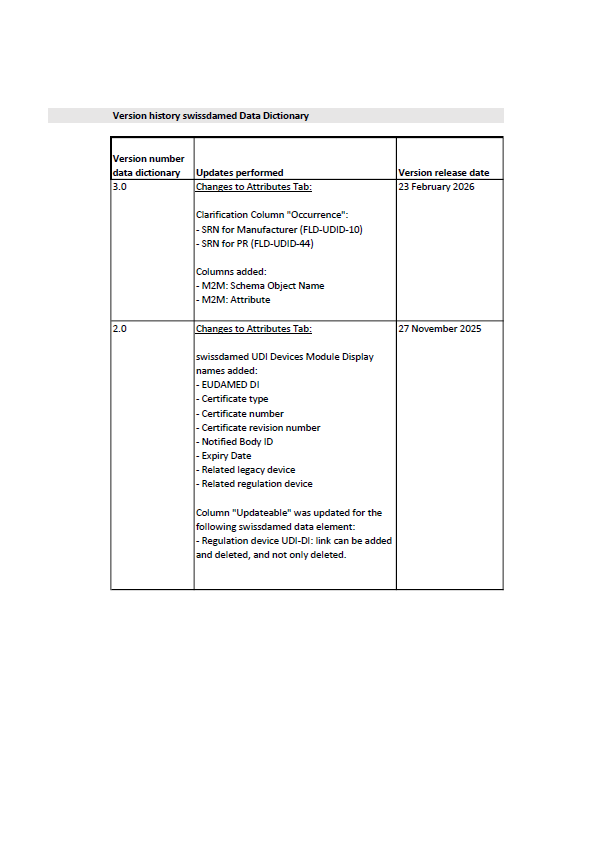
Swissmedic Updates swissdamed UDI Data Dictionary
Swissmedic has published version 3.0 of the swissdamed UDI Data Dictionary, introducing clarifications to SRN fields and new machine-to-machine attributes for improved data mapping.

Swissmedic Publishes swissdamed Machine-to-Machine REST API Documentation (Version 1.0)
Swissmedic publishes Version 1.0 of the swissdamed Machine-to-Machine REST API documentation for UDI registration and market status management.

ANVISA Publishes Draft Normative Instruction on UDI Data Transmission and SIUD Management
ANVISA publishes draft rules on UDI data transmission and SIUD management in Brazil. Learn how the new requirements impact medical device manufacturers.
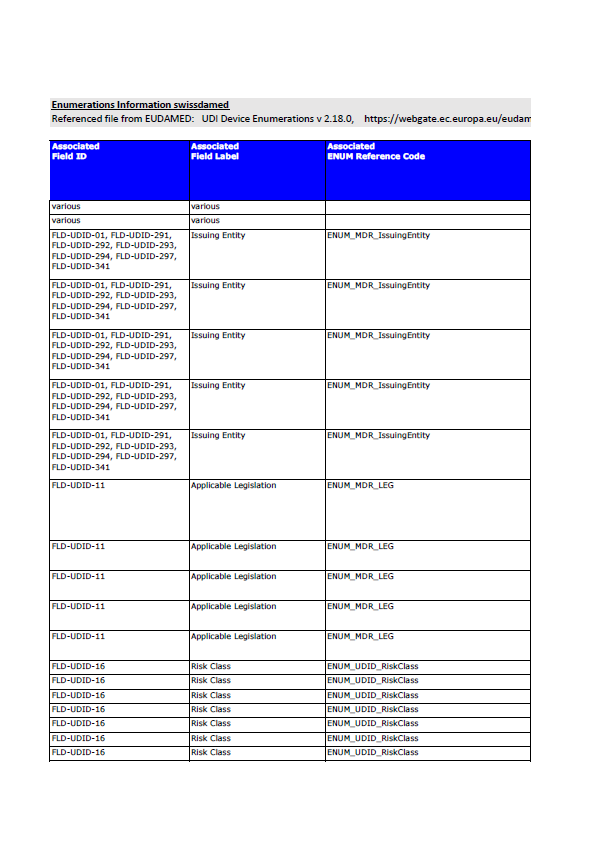
Swissmedic Publishes Updated EUDAMED UDI Enumerations: What Manufacturers Should Know
Swissmedic publishes updated EUDAMED UDI enumerations (v2.18.0). What this means for manufacturers preparing UDI, device and certificate data submissions.

Swissmedic Updates swissdamed Business Rules – Key Changes for Device Manufacturers in 2026
Swissmedic has released version 3.0 of its swissdamed Business Rules, effective January 2026. The update includes stricter UDI-DI requirements, limits on legacy device data, and mandatory fields for MDR/IVDR submissions. Manufacturers must now align with Swiss-specific data rules diverging from EUDAMED.

ANVISA publishes final version (v1.3) of the Medical Device Registration Manual – January 2026
ANVISA publishes final version (v1.3) of its Medical Device Registration Manual. See what manufacturers need to know for regulatory submissions in Brazil.

EUDAMED v2.18.0: Nova Atualização Traz Melhorias Importantes para Fabricantes e Distribuidores de Dispositivos Médicos
A EUDAMED é a base de dados europeia que centraliza informação sobre dispositivos médicos e de diagnóstico in vitro (IVD), essencial para a conformidade com o Regulamento (UE) 2017/745 (MDR) e o Regulamento (UE) 2017/746 (IVDR).
Com a atualização v2.18.0, fabricantes, distribuidores e representantes autorizados beneficiam de um sistema mais robusto, transparente e interoperável.