
FDA Finalizes Risk-Based Guidance on Human Factors Information in Medical Device Submissions
The FDA has published its final guidance on Human Factors information in medical device submissions, introducing a new risk-based framework and three Human Factors Submission Categories. Learn what manufacturers need to know.

FDA Replaces 25-Year-Old Bioequivalence Statistical Guidance with Major Update
The FDA has released a major update to its Bioequivalence Statistical Guidance, replacing the 2001 version. The new document introduces adaptive designs, modern statistical methods, guidance for highly variable drugs, NTI products, missing data handling and model-based approaches for generic drug development.

Australia Updates Guidance on UDI Compliance Timeframes for Medical Devices
The TGA has updated its guidance on UDI compliance timeframes for medical devices in Australia, including clarifications on legacy devices, EU MDR transitions, consignment stock, and Surgical Loan Kits.

Proposed MDR Article 50 Raises Concerns Over Notified Body Independence and Financial Sustainability
A new legal opinion analyses the European Commission’s proposed MDR Article 50, highlighting potential impacts on notified bodies, SMEs, certification costs, and conformity assessment access.

ANVISA Publishes 2024–2025 Good Clinical Practice Inspection Metrics: What Clinical Trial Sponsors and Manufacturers Should Know
ANVISA’s new 2024–2025 GCP inspection report highlights critical findings in clinical trials, data integrity, electronic systems, SOPs, and investigational product management. Learn what manufacturers and sponsors should review.

Swissmedic Highlights Updated MIR Template for Serious Incident Reporting Under MDR/IVDR
Swissmedic highlights the updated MIR template version 7.3.1 for MDR/IVDR vigilance reporting. Learn how the new requirements impact medical device and IVD manufacturers.

TGA Introduces Streamlined UDI Consent-to-Supply Pathway for Non-Compliant Medical Devices
Australia’s TGA has released new guidance on streamlined UDI Consent-to-Supply applications for medical devices that do not meet UDI-related Essential Principles. Learn how the changes affect manufacturers, ARTG entries, timelines, fees, and compliance strategies.

European Commission Publishes Updated EUDAMED UDI Devices User Guide
The European Commission released version 2.27.0 of the EUDAMED UDI Devices User Guide, clarifying UDI registration, EMDN management, packaging levels, and lifecycle updates under MDR and IVDR.

HTA Coordination Group Adopts Guiding Principles on Data Transparency
The EU HTA Coordination Group has adopted new guiding principles on data transparency, clarifying confidentiality expectations for Joint Clinical Assessments involving medical devices and IVDs.

TGA Expands Recognition of UK Approved Bodies for Medical Devices and IVDs
The Australian TGA has updated its guidance to recognise UK Approved Bodies as comparable overseas regulators, potentially simplifying ARTG submissions for medical device and IVD manufacturers.


FDA Announces New eMDR System Enhancements: What Medical Device Manufacturers Should Know
The FDA has announced new eMDR system enhancements, including AEMS integration updates, country code validation changes, and IMDRF adverse event code updates impacting medical device manufacturers.
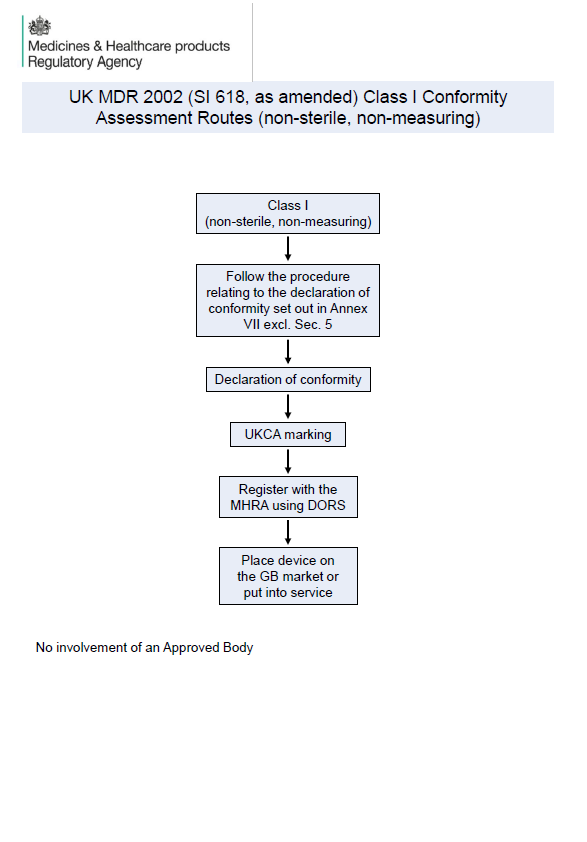
MHRA Publishes Visual Guidance on UK MDR 2002 Conformity Assessment Routes
The MHRA has released new visual guidance on UK MDR 2002 conformity assessment routes for medical devices. Learn what Class I, IIa, IIb, III, AIMD, and custom-made device manufacturers need to know for UKCA compliance and GB market access.

TGA Updates Guidance on the Use of Comparable Overseas Regulatory Assessments for Medical Devices
The Australian TGA updated its guidance on the use of overseas regulatory assessments for medical devices, including recognition of UK MHRA approved bodies. Learn what this means for manufacturers seeking ARTG inclusion.

European Commission Adopts New Rules for Notified Body Timelines, Quotations and Re-Certification Under MDR and IVDR
The European Commission has adopted Regulation (EU) 2026/977 introducing new MDR and IVDR rules for notified body timelines, quotations, transparency and re-certification activities.
Swissmedic Updates swissdamed Actors User Guide: What Manufacturers and Authorised Representatives Should Know
Swissmedic has updated the swissdamed Actors User Guide (v2.0), introducing AGOV login migration updates, mandate transfer functionality, and clarified actor management requirements for medical device manufacturers and authorised representatives in Switzerland.

FDA Releases New Guidance on Postapproval Pregnancy Safety Studies for Drugs and Biologics
The FDA has released new guidance on postapproval pregnancy safety studies for drugs and biologics, highlighting pregnancy registries, real-world evidence, and postmarketing surveillance expectations for manufacturers.

EU Reaches Provisional Agreement to Simplify AI Act Implementation
The EU Council and Parliament reached a provisional agreement to simplify the AI Act, including delayed deadlines for high-risk AI systems and clarifications for manufacturers.

New EU Implementing Regulation Introduces Greater Predictability for Conformity Assessment
The European Commission has adopted Implementing Regulation (EU) 2026/977, introducing new conformity assessment requirements for notified bodies under the MDR and IVDR.

European Commission Updates MIR 7.3.1 Vigilance Reporting Forms
The European Commission has updated the MIR 7.3.1 vigilance reporting forms under the EU MDR and IVDR. Manufacturers should verify accepted versions and updated XSD/XSL files.