MHRA Publishes Visual Guidance on UK MDR 2002 Conformity Assessment Routes
The UK Medicines and Healthcare products Regulatory Agency (MHRA) has published a new visual guidance document summarising the conformity assessment routes under the UK Medical Devices Regulations 2002 (UK MDR 2002, SI 618 as amended).
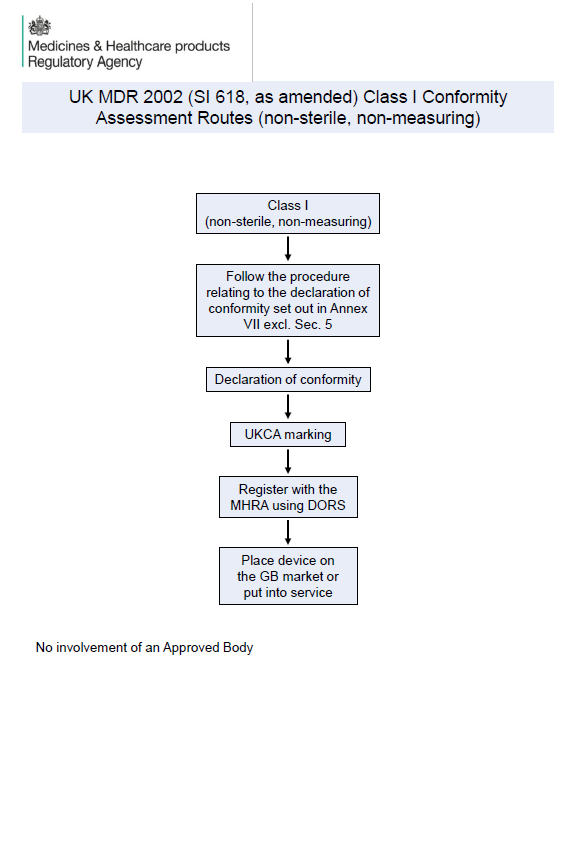
The document provides flowchart-based overviews for multiple device categories, including:
Class I devices (non-sterile, sterile, and measuring function)
Class IIa, IIb, and III devices
Systems and procedure packs
Custom-made devices
Active implantable medical devices (AIMDs)
The guidance outlines the applicable conformity assessment procedures, the role of UK Approved Bodies, UKCA marking requirements, and MHRA registration obligations through the DORS system.
Why this matters for manufacturers
Although the document does not introduce new regulatory requirements, it provides a consolidated and simplified overview of the existing UK conformity assessment pathways. This is particularly relevant for manufacturers currently:
Transitioning from CE marking strategies to UKCA compliance
Planning market access in Great Britain
Assessing the level of Approved Body involvement
Reviewing regulatory responsibilities for Class I sterile or measuring devices
Managing custom-made devices or procedure packs
For many manufacturers, one of the most important clarifications is the distinction between:
Class I non-sterile/non-measuring devices, which do not require Approved Body involvement; and
Class Is/Im devices, where Approved Body review is limited to sterility or metrological aspects.
The guidance also reinforces that:
Manufacturers must register devices with the MHRA using DORS
UKCA marking must include the Approved Body number where applicable
Custom-made devices must not bear a UKCA mark
Procedure packs rely on compliance of the individual constituent devices
Strategic considerations
Manufacturers placing devices on the GB market should carefully verify whether their current conformity assessment route remains appropriate under UK MDR 2002 requirements.
The publication may also help regulatory teams identify:
Potential gaps in technical documentation
Incorrect assumptions regarding Approved Body involvement
Device classification-related risks
Registration and declaration of conformity obligations
Companies using EU MDR-based processes should remember that the UK system continues to rely on the legacy UK MDR 2002 framework, which is structurally aligned with the former EU directives rather than the EU MDR.
Read the full document below.