
Swissmedic Updates Guidance on Export Certificates for Medical Devices
Swissmedic has updated its guidance on export certificates for medical devices, including eligibility, ordering requirements, product lists, CE conformity documentation, swissdamed registration and fees.

TGA Updates Guidance on Selection Criteria for Medical Device Application Audits
The TGA has updated its guidance on how medical device applications are selected for audit before inclusion in the ARTG, including mandatory audits, non-mandatory audit criteria, regulatory reforms, UDI and post-market signals.

Team-NB Highlights Risk-Adaptive Surveillance under the MDR and IVDR
Team-NB has presented a proposal for risk-adaptive notified body surveillance under the MDR and IVDR, aiming to reduce unnecessary burden while maintaining patient safety and effective oversight.
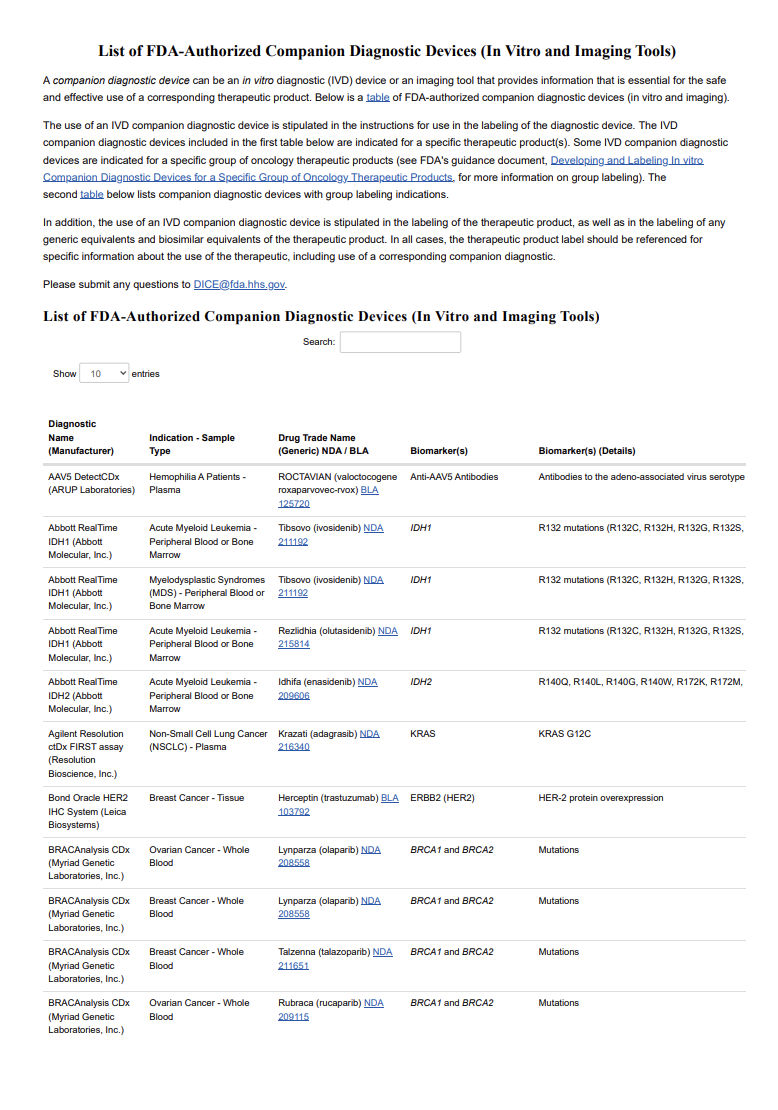
FDA Updates Authorized Companion Diagnostic Devices List to 226 Entries
FDA updates its list of authorized companion diagnostic devices to 226 entries, covering NGS assays, liquid biopsies, group labeling, and key biomarkers.

MHRA Releases AI Airlock Sandbox Phase 2 Programme Report
MHRA releases its AI Airlock Sandbox Phase 2 report, detailing regulatory insights on AI validation, PCCPs, generative AI, and real-world PMS.

European Commission Renews UDI Issuing Entity Designations Until 2029
Commission Implementing Decision (EU) 2024/2120 renews the designation of UDI issuing entities for medical devices and IVDs for a further five-year period, until 27 June 2029.

MDCG Publishes Position Paper on UDI Assignment Between Manufacturers and Distributors
MDCG 2026-5 clarifies that only the manufacturer can assign UDIs to devices placed on the EU market, even where distributors market devices under their own brand.

European Commission Publishes 2025 Member State Summaries on Notified Body Monitoring
The European Commission has published Member State summaries of the 2025 annual reports on monitoring and on-site activities regarding notified bodies under the MDR and IVDR.

Swissmedic Updates Information Sheet on IVD Performance Studies
Swissmedic has updated its information sheet on performance studies of IVDs, covering study categories, approval processes, ISO 20916, reporting duties, safety measures and sponsor obligations.

MHRA Updates Guidance on Registering Medical Devices for the UK Market
The MHRA has updated its guidance on registering medical devices, IVDs, custom-made devices, systems and procedure packs for the Great Britain and Northern Ireland markets.
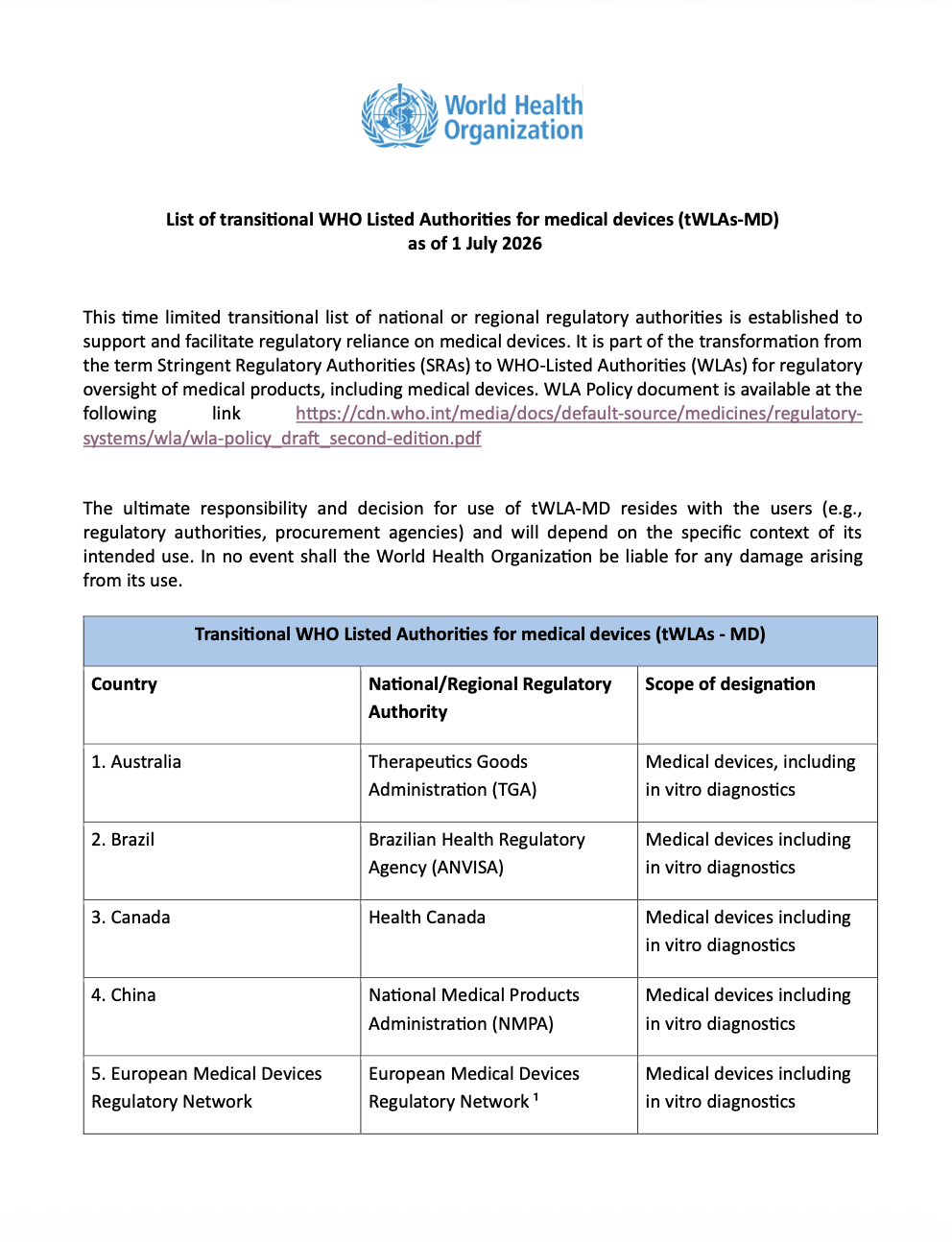
WHO Publishes Transitional List of WHO Listed Authorities for Medical Devices
The World Health Organization has published a transitional list of WHO Listed Authorities for medical devices, supporting regulatory reliance for medical devices and IVDs.

Swissmedic Updates Checklist for Materiovigilance Inspections in Hospitals
Swissmedic has updated its checklist for materiovigilance inspections in hospitals, covering medical devices and IVDs, quality management, reporting, traceability, FSN/FSCA processes and staff training.

MHRA Updates Guidance on IVD Point-of-Care Test Devices
The MHRA has updated its guidance on the management and use of in vitro diagnostic point-of-care test devices, including POCT services in primary and secondary care settings.

MedTech Europe Responds to European Commission Call for Evidence on Joint Undertakings
MedTech Europe has responded to the European Commission’s Call for Evidence on future European Partnerships implemented as Joint Undertakings under the next Multiannual Financial Framework.

MHRA Updates List of UK Approved Bodies for Medical Devices
The MHRA has updated its list of UK approved bodies authorised to undertake conformity assessments for medical devices under the UK MDR 2002.

TGA UDI Requirements for Medical Devices Commence in Australia
From 1 July 2026, certain medical devices supplied in Australia must meet TGA Unique Device Identification requirements, starting with Class IIb and Class III devices.

NBCG-MED Publishes Operational Elements for Hybrid Audits under MDR and IVDR
NBCG-MED has published operational elements for the application of hybrid audits to QMS assessments under the MDR and IVDR, including on-site requirements, ICT use and audit planning.

European Commission Drafts Updated Common Specifications for Certain Class D IVDs
The European Commission has published a draft implementing regulation amending common specifications for certain Class D IVDs under the IVDR, including HEV, Toxoplasma gondii, Plasmodium and arboviruses.

Swissmedic Updates Information Sheet on IVD Performance Studies
Swissmedic has updated its information sheet on IVD performance studies, covering approval procedures, reporting duties, ISO 20916, category C studies and Swissmedic surveillance.

MDCG Updates Guidance on Standardisation for Medical Devices
The MDCG has updated its guidance on standardisation for medical devices, covering harmonised standards, presumption of conformity, state of the art, common specifications and EU case law.